|
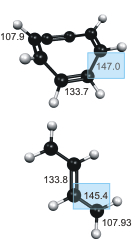
We combine femtosecond time-resolved rotational coherence spectroscopy with high-level ab initio theory to obtain accurate structural information for the nonpolar antiaromatic molecule 1,3,5,7-cyclooctatetraene (C8H8, COT) and its perdeuterated isotopomer COT-d8 (C8D8). The experimental rotational and centrifugal distortion constants are compared to those obtained from calculations at the coupled-cluster with single, double, and perturbative triples [CCSD(T)] level. The latter also take into account vibrational averaging effects of the ground and vibrationally excited states. Combining the experimental and calculated rotational constants with the calculated equilibrium bond lengths and angles allows us to determine accurate equilibrium structure parameters, e.g., re(C-C) = 147.0 ± 0.05 pm, re(C=C) = 133.7 ± 0.1 pm, and re(C-H) = 107.9 ± 0.1 pm. The equilibrium C-C and C=C bond lengths of COT are compared to those of 1,3-butadiene. The expected effect of decreased π-electron delocalization due to the twisting of adjacent C=C double bonds in COT relative to butadiene is observed for the C-C bonds but not for the C=C bonds.
This work was carried out in the group of Prof. Samuel Leutwyler.
References:
-
D. S. Kummli, S. Lobsiger, H. M. Frey, S. Leutwyle, J. F. Stanton;
"Accurate Determination of the Structure of Cyclooctatetraene by Femtosecond Rotational Coherence Spectroscopy and ab Initio Calculations"
J. Phys. Chem. A, 112, 9134-9143, (2008);
doi:10.1021/jp803523y.
|